Awesome
<!--brand: |- <a href="http://flow-r.github.io/flowr"> <img src='https://raw.githubusercontent.com/sahilseth/flowr/devel/vignettes/files/logo.png' alt='flowr icon' width='40px' height='40px' style='margin-top: -10px;'> </a> --> <meta property="og:description" content="Easy, scalable big data pipelines using hpcc (high performance computing cluster)"> <meta property="og:title" content="flowr — Easy, scalable big data pipelines using hpcc"> <meta name="twitter:description" content="flowr - Easy, scalable big data pipelines using hpcc (high performance computing cluster)"> <meta name="twitter:title" content="flowr — Easy, scalable big data pipelines using hpcc (high performance computing cluster)">![]()
 Streamlining Computing workflows
Streamlining Computing workflows
Latest documentation: flow-r.github.io/flowr
Flowr framework allows you to design and implement complex pipelines, and deploy them on your institution's computing cluster. This has been built keeping in mind the needs of bioinformatics workflows. However, it is easily extendable to any field where a series of steps (shell commands) are to be executed in a (work)flow.
Highlights
- No new syntax or language. Put all shell commands as a tsv file called flow mat.
- Define the flow of steps using a simple tsv file (serial, scatter, gather, burst...) called flow def.
- Works on your laptop/server or cluster (/cloud).
- Supports multiple cluster computing platforms (torque, lsf, sge, slurm ...), cloud (star cluster) OR a local machine.
- One line installation (
install.packages("flowr")) - Reproducible and transparent, with cleanly structured execution logs
- Track and re-run flows
- Lean and Portable, with easy installation
- Fine grain control over resources (CPU, memory, walltime, etc.) of each step.
Example
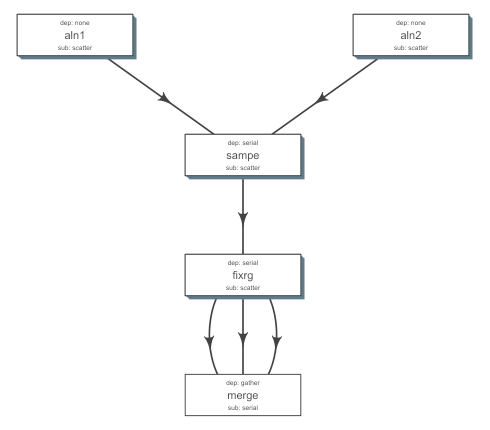
A few lines, to get started
## Official stable release from CRAN (updated every other month)
## visit flow-r.github.io/flowr/install for more details
install.packages("flowr", repos = "http://cran.rstudio.com")
# or a latest version from DRAT, provide cran for dependencies
install.packages("flowr", repos = c(CRAN="http://cran.rstudio.com", DRAT="http://sahilseth.github.io/drat"))
library(flowr) ## load the library
setup() ## copy flowr bash script; and create a folder flowr under home.
# Run an example pipeline
# style 1: sleep_pipe() function creates system cmds
flowr run x=sleep_pipe platform=local execute=TRUE
# style 2: we start with a tsv of system cmds
# get example files
wget --no-check-certificate http://raw.githubusercontent.com/sahilseth/flowr/master/inst/pipelines/sleep_pipe.tsv
wget --no-check-certificate http://raw.githubusercontent.com/sahilseth/flowr/master/inst/pipelines/sleep_pipe.def
# submit to local machine
flowr to_flow x=sleep_pipe.tsv def=sleep_pipe.def platform=local execute=TRUE
# submit to local LSF cluster
flowr to_flow x=sleep_pipe.tsv def=sleep_pipe.def platform=lsf execute=TRUE
Example pipelines inst/pipelines
Resources
- For a quick overview, you may browse through, these introductory slides.
- The overview provides additional details regarding the ideas and concepts used in flowr
- We have a tutorial which can walk you through creating a new pipeline
- Additionally, a subset of important functions are described in the package reference page
- You may follow detailed instructions on installing and configuring
- You can use flow creator: https://sseth.shinyapps.io/flow_creator), a shiny app to aid in designing a shiny new flow. This provides a good example of the concepts
Updates
This package is under active-development, you may watch for changes using the watch link above.
Feedback
Please feel free to raise a github issue with questions and comments.
Acknowledgements
- Jianhua Zhang
- Samir Amin
- Roger Moye
- Kadir Akdemir
- Ethan Mao
- Henry Song
- An excellent resource for writing your own R packages: r-pkgs.org