Awesome
DEPRECATED
This sofwtare is outdated.
VIZBAM
Java library displaying SAM / BAM alignments.
Project status: beta
Author: Pierre Lindenbaum PhD. @yokofakun
Dependencies
- Java 1.7
- Picard http://picard.sourceforge.net/
- apache ant
Screenshots
Displaying a BAM like samtools tview (part of the project https://github.com/lindenb/ngsproject )
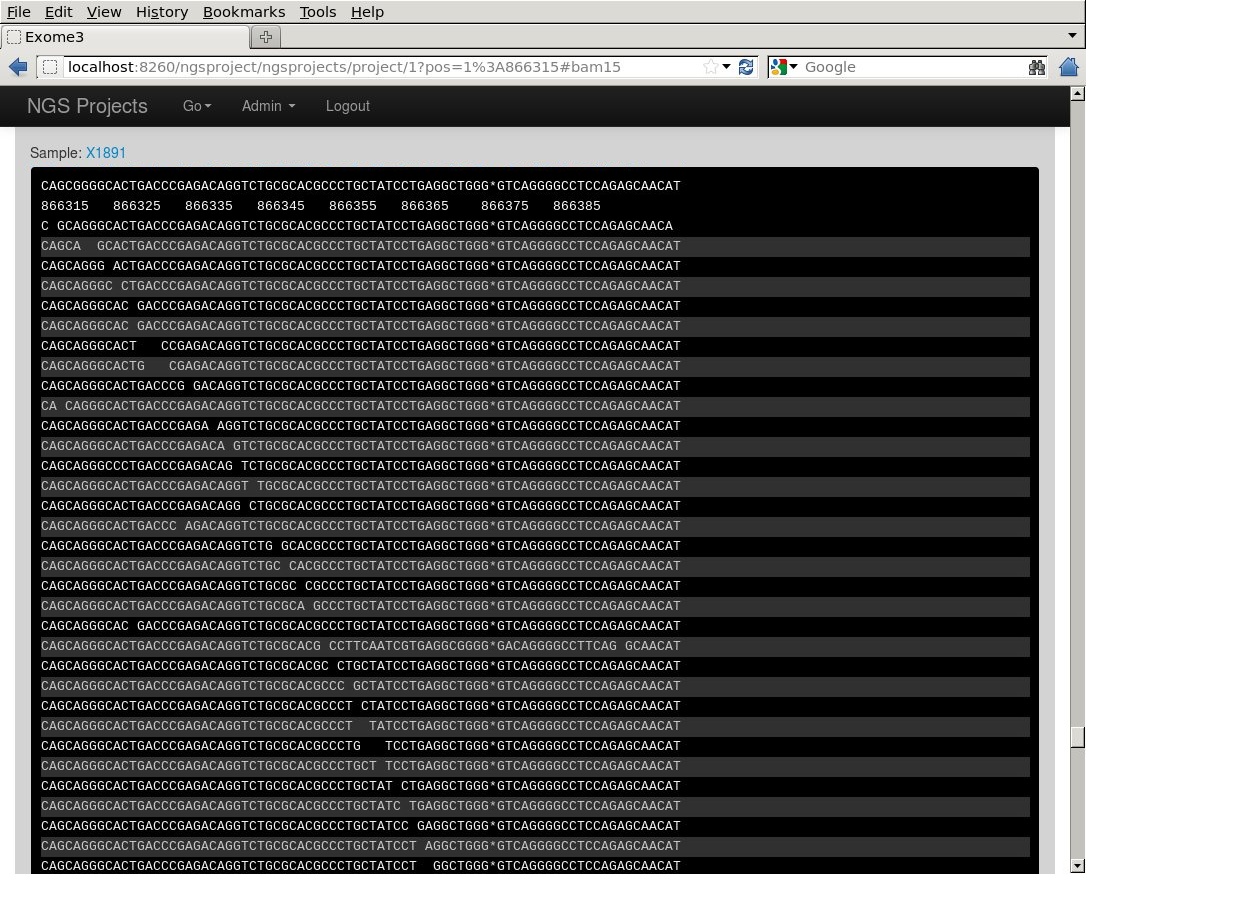
Cmd-Line Tools
<h3>VizBamCmdLine</h3> <p>USAGE: VizBamCmdLine [options] <p> <p>Print a SAM alignment like samtools tview</p> <table> <tr><th>Option</th><th>Description</th></tr> <tr><td>FORMAT=String</td><td>output format Default value: text. This option can be set to 'null' to clear the default value. </td></tr> <tr><td>REFERENCE=File</td><td>Input indexed reference Required. </td></tr> <tr><td>OUT=File</td><td>out file Default value: null. </td></tr> <tr><td>INPUT=File</td><td>A BAM file to process. Required. </td></tr> <tr><td>POSITION=String</td><td>The position to process. Syntax is "chrom:position" the chromosome must be present in the reference . Default value: null. </td></tr> <tr><td>INTERVALFILE=File</td><td>File containing intervals. Default value: null. </td></tr> <tr><td>WIDTH=Integer</td><td>The screen width. Default value: 80. This option can be set to 'null' to clear the default value. </td></tr> <tr><td>USECLIPPED=Boolean</td><td>Use Clipped Ends. Default value: false. This option can be set to 'null' to clear the default value. Possible values: {true, false} </td></tr> <tr><td>BASE_QUALITY=Boolean</td><td>Handle Base quality Default value: false. This option can be set to 'null' to clear the default value. Possible values: {true, false} </td></tr> </table> <br/>Installation
git clone "https://github.com/lindenb/vizbam.git"
cd vizbam.git
edit build.properties to configure the project. Something like:
picard.version=1.91
picard.dir=/home/lindenb/package/picard/picard-tools-${picard.version}
compile the library
ant