Awesome
Imputation Workflow h3abionet/chipimputation
![]()
Introduction
Imputation is likely to be run in the context of a GWAS, studying population structure, and admixture studies. It is computationally expensive in comparison to other GWAS steps. The basic steps of the pipeline is described in the diagram below:
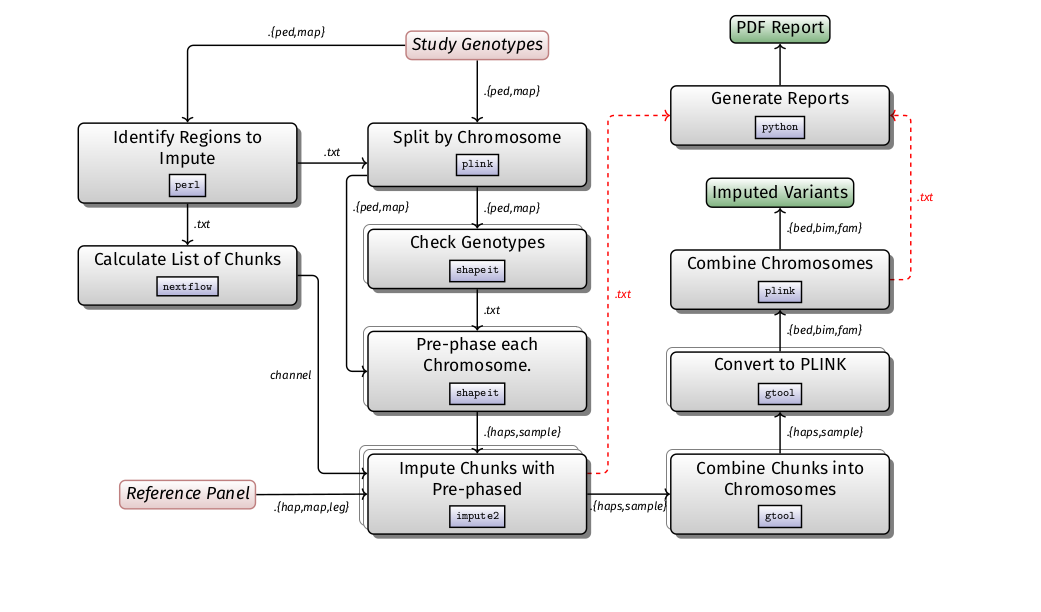
- The workflow is developed using
 and imputation performed using Minimac4.
and imputation performed using Minimac4. - It identifies regions to be imputed on the basis of an input file in VCF format, split the regions into small chunks, phase each chunk using the phasing tool Eagle2 and produces output in VCF format that can subsequently be used in a GWAS workflow.
- It also produce basic plots and reports of the imputation process including the imputation performance report, the imputation accuracy, the allele frequency of the imputed vs of the reference panel and other metrics.
This pipeline comes with docker/singularity containers making installation trivial and results highly reproducible.
Getting started
Running the pipeline with test dataset
This pipeline itself needs no installation - NextFlow will automatically fetch it from GitHub. You can run the pipeline using test data hosted in github with singularity without have to install or change any parameters.
nextflow run h3abionet/chipimputation/main.nf -profile test,singularity
testprofile will download the testdata from heresingularityprofile will download the singularity image from quay registry
Check for results in ./output
Start running your own analysis
Copy the test.config file from the conf folder by doing cp <conf dir>/test.config . and edit it to suit the path to where your files are stored.
Once you have edited the config file, run the command below.
nextflow run h3abionet/chipimputation -c "name of your config file" -profile singularity
singularityprofile will download the singularity image from quay registry
Check for results in ./output
Documentation
The h3achipimputation pipeline comes with detailed documentation about the pipeline.
This is found in the docs/ directory:
- Installation
- Pipeline configuration
2.1. Configuration files
2.2. Software requirements
2.3. Other clusters - Running the pipeline with test data
- Running the pipeline with your own config
- Running on local machine or cluster
- Running docker and singularity
Support
We track our open tasks using github's issues
Citation
This workflow which was developed as part of the H3ABioNet Hackathon held in Pretoria, SA in 2016. Should want to reference it, please use:
Baichoo S, Souilmi Y, Panji S, Botha G, Meintjes A, Hazelhurst S, Bendou H, Beste E, Mpangase PT, Souiai O, Alghali M, Yi L, O'Connor BD, Crusoe M, Armstrong D, Aron S, Joubert F, Ahmed AE, Mbiyavanga M, Heusden PV, Magosi LE, Zermeno J, Mainzer LS, Fadlelmola FM, Jongeneel CV, Mulder N. Developing reproducible bioinformatics analysis workflows for heterogeneous computing environments to support African genomics. BMC Bioinformatics. 2018 Nov 29;19(1):457. doi: 10.1186/s12859-018-2446-1. PubMed PMID: 30486782; PubMed Central PMCID: PMC6264621.