Awesome
paraMSA
paraMSA incorporates the information from uncertainty in multiple sequence alignments into the construction of phylogenetic trees and the bootstrap support values. The application generates, combines and evaluates alternative multiple sequence alignments to provide more accurate phylogenetic trees and more descriminative support values.
Alternative alignments are generated through use of different guide trees, alignment orders and gap penalties using the CLUSTALW, MAFFT and PRANK progressive aligners. The alternative alignments are then applied to phylogenetic reconstruction through generation of phylogenetic trees and support trees.
A new phylogenetic tree support value termed the 'parastrap' is presented. The support value is derived from any number of alternative alignments which are in turn bootstrapped using the traditional bootstrap method to create parastrap replicates. Each parastrap replicate can then be used to create a create a replicate tree. Tree and node evaluation versus a known phylogenetic tree is performed using the T-Coffee seq-reformat tools.
Schematic Outline
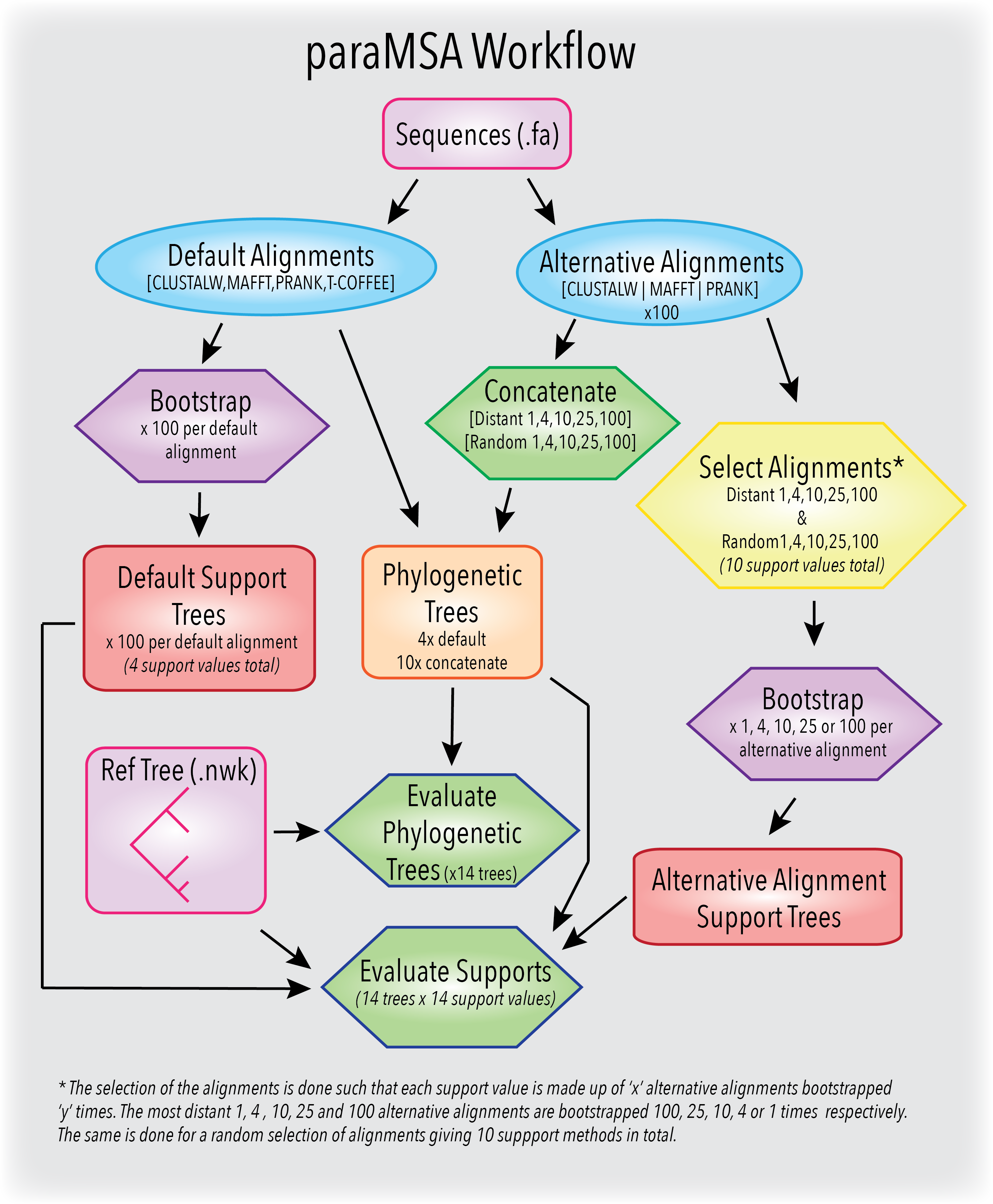
You can find the Nextflow generated directed acyclic graph here
Quick start
Make sure you have all the required dependencies listed in the last section.
Install the Nextflow runtime by running the following command:
$ curl -fsSL get.nextflow.io | bash
When done, you can launch the pipeline execution by entering the command shown below:
$ nextflow run skptic/paraMSA
By default the pipeline is executed against the provided example dataset. Check the Pipeline parameters section below to see how enter your data on the program command line.
Pipeline parameters
--seq
- Specifies the location of the fasta file(s) containing sequences.
- Multiple files can be specified using the usual wildcards (*, ?), in this case make sure to surround the parameter string value by single quote characters (see the example below)
- By default it is set to the paraMSA location:
./tutorial/data/tips16_0.5_00{1,2}.0400.fa
Example:
$ nextflow run skptic/paraMSA --seq '/home/my_dataset/*.fasta'
This will handle each fasta file as a seperate alignment/tree/bootstrap support
--ref
- Specifies the reference phylogentic tree in NEWICK format
- This is used for the evaluation of phylogentic trees and support values
Example:
$ nextflow run skptic/paraMSA --ref '/home/my_dataset/referenceTree.nwk'
--aligner
-
Specifies the aligner to use for the generation of alternative alignments
-
Options are
CLUSTALW,MAFFTandPRANK$ nextflow run skptic/paraMSA --aligner 'MAFFT'
--alignments
- Specifies the number of alignments to sample from the guidance2 alternative alignments.
- Min = 1, Max = 400, MUST BE MULTIPLE OF 4
- By default it is set to
16
Example:
$ nextflow run skptic/paraMSA --alignments '32'
--straps
- Specifies the number of support replicates to create per alternative alignment
- By default it is set to
16
Example:
$ nextflow run skptic/paraMSA --straps '100'
NOTE: The total number of parastrap replicates generated is equal to the number of alternative alignments multiplied by the number of bootstraps. Given the defaults of 16 alginments and 16 bootstraps then 256 support replicates/trees are generated.
--alignments_list
- A list specifing the number of alignments to use for both phylogenetic trees and support values.
- By default this is set to
1,2,4,8,16 - This equates to 5 phylogenetic trees being generated, each constructed from the concatenation of either 1, 2, 4, 8 or 16 alternative alignments.
- The largest number in the list must be equal to or less that the number specified by
--alignments
Example:
$ nextflow run skptic/paraMSA --alignments_list '1,10,50,100'
--straps_list
- A list specifing the number of bootstraps to use for the support values.
- By default this is set to
1,2,4,8,16 - For each of the numbers in the
--alignments_list, a support value is created by the bootstraping this number of alignments by the number times specified in the--straps_list. - For the default values this result in support values from 1-1, 1-2, 1-4, 1-8, 1-16, 2-1, 2-2, 2-4...etc where 1-1 is one alternative alignment bootstrapped one time and 2-4 is two alternative alignments bootstrapped twice (to give four support replicates)
- The largest number in the list must be equal to or less that the number specified by
--straps
Example:
$ nextflow run skptic/paraMSA --alignments_list '1,10,50,100'
--output
- Specifies the folder where the results will be stored for the user.
- It does not matter if the folder does not exist.
- By default is set to paraMSA's folder:
./tutorial/results
Example:
$ nextflow run skptic/paraMSA --output /home/user/my_results
--name
- Specifies the name of the execution
- By default is set to:
tutorial_data
Example:
$ nextflow run skptic/paraMSA --name `my_run_001`
Cluster support
concTree-NF execution relies on Nextflow framework which provides an abstraction between the pipeline functional logic and the underlying processing system.
Thus it is possible to execute it on your computer or any cluster resource manager without modifying it.
Currently the following platforms are supported:
- Oracle/Univa/Open Grid Engine (SGE)
- Platform LSF
- SLURM
- PBS/Torque
By default the pipeline is parallelized by spanning multiple threads in the machine where the script is launched.
To submit the execution to a SGE cluster create a file named nextflow.config, in the directory
where the pipeline is going to be launched, with the following content:
process {
executor='sge'
queue='<your queue name>'
}
In doing that, tasks will be executed through the qsub SGE command, and so your pipeline will behave like any
other SGE job script, with the benefit that Nextflow will automatically and transparently manage the tasks
synchronisation, file(s) staging/un-staging, etc.
Alternatively the same declaration can be defined in the file $HOME/.nextflow/config.
To lean more about the avaible settings and the configuration file read the Nextflow documentation.
Dependencies
- Java 7+
- Guidance2
- ClustalW2
- MAFFT
- PRANK
- T-Coffee
- FastTree
- HH Suite
- Easal Tools (from HMMER)
- Algorithm::Cluster (Perl Module)