Awesome
Zetadesign
There is CodeOcean link: (https://codeocean.com/capsule/5271163/tree/v1)
Author Email: joreyyan@buaa.edu.cn
Publication: https://doi.org/10.1093/bib/bbad257
Environment
You can configure the environment using either requirements.txt or conda+environment.yaml. It is recommended to use the latter as some packages have specific requirements.
For example, pdbfixer is recommended to be installed using conda install -c conda-forge pdbfixer as it requires the latest version of pdbfixer which uses openmm8.0.
However, this may cause conflicts with other packages, so please be cautious during installation. It is recommended to use Python version 3.9 or lower to avoid potential errors with the OpenMM version exceeding 7.7. If you encounter any issues, feel free to contact me via email.
Data
We use the cath4.3-s40-no redundant dataset, which can be downloaded using the multi-threaded script /data/download-chains.py. Alternatively,
you can directly download the complete files from [ftp]ftp://orengoftp.biochem.ucl.ac.uk/cath/releases/latest-release/non-redundant-data-sets/.
We filter the original files using /data/cath/spilt_dataset.py, for example by applying a resolution threshold of <3 Å,
and then split the dataset into training and test sets following the 95% - 5% ratio described in the paper. This process generates a final text file with the dataset configuration.
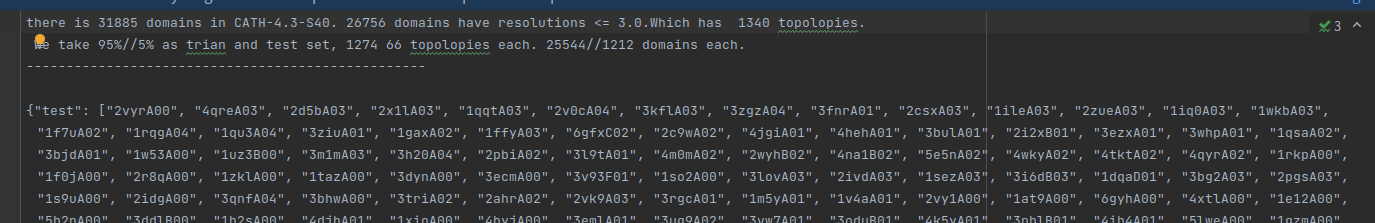
Some PDB files (approximately a few dozen) have insert code errors. You can choose to drop them or use pdb-tools to correct them. Our spilt_dataset.py also includes the write_trainandtest method to output PDB files as JSON. Additionally, you can use the test_insert() method in single_chain_split.py to identify which PDB files have issues.
We provide several methods in single_chain_split.py to write PDB files as pickle files, each with a different amount of extracted features.
If you want to train the Fix Backbone Design algorithm, we recommend using the write_fbb_pdb_files_to_pickl method. For training the Repacking algorithm, you can use write_allatomspdb_files_to_pickl. The length of each training data will be truncated to 256. If you are running FBB design or repacking on multiple new PDB files, you can use write_design_pdb_files_to_pickl to write them as a single pickle file. Please note that this method requires the installation of DSSP tool, which can be installed beforehand from https://swift.cmbi.umcn.nl/gv/dssp/.
If you want to test new sample PDB files, you can place them in the /demo/ folder. After running single_chain_split.py, you will obtain the processed_pdb.pkl file.
Note: If you run single_chain_split.py, you will need to have dssp installed locally, for example, by running conda install -c salilab dssp.
The PDB files used for testing in CASP14/CASP15 mentioned in the paper are located in the CASP14/CASP15 pkl folders.
Inference
For single_chain_split.py
Input flags:
# Add arguments for input and output directories
parser.add_argument('--input_dir', type=str, default='./demo/', help='Path to input PDB directory')
parser.add_argument('--output_dir', type=str, default='./demo/', help='Path to output pkl directory')
For Joint_to_repacker.py
Input flags:
# Add arguments for input and output directories
parser.add_argument('--use_amber', action='store_true', default=False, help='Using Amber for optimizing the output results')
parser.add_argument('--use_fbb', action='store_true', default=False, help='Using fbb to generate new sequences')
parser.add_argument('--pkl_dir', type=str, default='./design/CASP15_nocut.pkl', help='pkl file made from pdbs')
Example for Repacking Only/ to repack some pdbs:
python Joint_to_repacker.py --pkl_dir=''
Example for Repacking and use amber to relax result/ to repack some pdbs:
python Joint_to_repacker.py --use_amber --pkl_dir=''
Example for design new sequences, repacking and use amber to relax result/ to repack some pdbs:
python Joint_to_repacker.py --use_amber --user_fbb --pkl_dir=''
Others
If you want to use the repacker to process sequences generated by other methods, such as those generated by MPNN, you can use the design_byseq method in Joint_to_repacker.py. This method takes name and seq as inputs, where name is the name of the PDB file and seq is the newly generated sequence from another method. Both inputs should be strings.
To use this method, first preprocess the original PDB file into a pkl file. Then, modify the args.design(None, argss.use_fbb, argss.use_amber) line in the main function to args.design_byseq(None, name, seq).
Note: If you want to use design_byseq, you may need to make slight modifications to the print/log statements inside the method. However, you can refer to the design method as a reference.
logging
For Repacking Only/, The first line is used to print the predicted accuracy. The first three angles represent the backbone angle errors. When performing repacking, we also make slight modifications to the backbone atom positions, so the errors in these angles are very small. The next four angles represent the chi angle errors, which increase progressively.
98%|█████████▊| 44/45 [00:14<00:00, 2.07it/s]2023-05-19 13:45:45,893 ----------------------------------------------------------------------------------------------------
2023-05-19 13:45:45,893 designing T1180-D1
2023-05-19 13:45:46,389 ca_lddt:100.00;Angle_accs1:3.00;2:4.00;3:1.00;chi1:21.00;chi2:27.00;chi3:43.00;chi4:58.00;
2023-05-19 13:45:46,389 bfloss:1.82;fapeloss:3.09;chi_angle_loss:2.17;bb_angle_loss:0.01;step 0;
2023-05-19 13:45:46,389 --------------------------------------------------
2023-05-19 13:45:46,415 ----------------------------------------------------------------------------------------------------
For Repacking and use amber to relax result/ ,In addition to displaying the accuracy as shown above, the following part involves using Amber to perform relaxation on the repacked results. The relaxed structure is then saved as 'xxx_relaxed.pdb'.
4%|▍ | 2/45 [00:09<03:11, 4.46s/it]2023-05-19 13:51:28,987 ----------------------------------------------------------------------------------------------------
2023-05-19 13:51:28,987 designing T1170-D2
2023-05-19 13:51:29,101 ca_lddt:100.00;Angle_accs1:1.00;2:3.00;3:0.00;chi1:17.00;chi2:18.00;chi3:45.00;chi4:27.00;
2023-05-19 13:51:29,101 bfloss:1.07;fapeloss:2.21;chi_angle_loss:1.56;bb_angle_loss:0.00;step 0;
2023-05-19 13:51:29,101 --------------------------------------------------
2023-05-19 13:51:29,106 Running relaxation on /home/jorey/zetadesign/output///PDHS_T1170-D2_unrelaxed.pdb...
2023-05-19 13:51:29,482 alterations info: {'nonstandard_residues': [], 'removed_heterogens': set(), 'missing_residues': {}, 'missing_heavy_atoms': {}, 'missing_terminals': {<Residue 71 (THR) of chain 0>: ['OXT']}, 'Se_in_MET': [], 'removed_chains': {0: []}}
2023-05-19 13:51:29,567 Minimizing protein, attempt 1 of 100.
2023-05-19 13:51:29,699 Restraining 577 / 1143 particles.
2023-05-19 13:51:30,112 alterations info: {'nonstandard_residues': [], 'removed_heterogens': set(), 'missing_residues': {}, 'missing_heavy_atoms': {}, 'missing_terminals': {}, 'Se_in_MET': [], 'removed_chains': {0: []}}
2023-05-19 13:51:30,173 Iteration completed: Einit 28692.26 Efinal -1353.53 Time 0.30 s num residue violations 0 num residue exclusions 0
2023-05-19 13:51:30,560 alterations info: {'nonstandard_residues': [], 'removed_heterogens': set(), 'missing_residues': {}, 'missing_heavy_atoms': {}, 'missing_terminals': {<Residue 71 (THR) of chain 0>: ['OXT']}, 'Se_in_MET': [], 'removed_chains': {0: []}}
2023-05-19 13:51:30,691 Relaxation time: 1.5848838239908218
2023-05-19 13:51:30,691 Relaxed output written to /home/jorey/zetadesign/output///T1170-D2_relaxed.pdb...
2023-05-19 13:51:30,691 designed Relaxed output written to /home/jorey/zetadesign/output///T1170-D2_relaxed.pdb...
2023-05-19 13:51:30,691 ----------------------------------------------------------------------------------------------------
For design new sequences, repacking and use amber to relax result/ , The accuracy display section now includes an additional metric called "recovery." The chi angle values shown at this stage represent the differences between the chi angles of the repacked sequence and the original sequence. It's important to note that these results are not directly comparable and are provided for reference purposes only.
84%|████████▍ | 38/45 [00:16<00:02, 3.04it/s]2023-05-19 13:54:56,577 ---------------------------------------------------------
2023-05-19 13:54:56,577 designing T1137S5-D1
2023-05-19 13:54:56,833 ca_lddt:100.00;Angle_accs1:3.00;2:4.00;3:1.00;chi1:28.00;chi2:39.00;chi3:91.00;chi4:23.00;recovery:49.64
2023-05-19 13:54:56,833 bfloss:5.03;fapeloss:3.41;chi_angle_loss:3.21;bb_angle_loss:0.01;step 0;
2023-05-19 13:54:56,833 --------------------------------------------------
2023-05-19 13:54:56,842 ----------------------------------------------------------------------------------------------------
Reference
Thanks for